Schilder P., 1912]. Редкое заболевание, обусловленное неуклонно прогредиентным демиелинизирующим процессом, поражающим вещество головного мозга, его больших полушарий и мозжечка.
Характеризуется нарастанием деменции и расстройств речи, эпилептиформными припадками, очаговыми поражениями, в зависимости от локализации процесса, слуха и зрения, вплоть до полной глухоты и амавроза, образованием спастических параличей и появлением псевдобульбарных симптомов. Смерть в течение двух-трех лет при явлениях кахексии и децеребрационной ригидности.
Начало заболевания чаще всего в молодом и, особенно, в детском возрасте, хотя не исключена возможность заболевания и в другие возрастные периоды.
Этиология неизвестна. Предположительно указывают на близость между Ш.б. и рассеянным склерозом.
Син.: диффузный склероз нервной системы, прогрессирующая мозговая лейкопатия, диффузный прогрессирующий склеротический периаксиальный энцефалит, прогрессирующая склеротическая энцефалолейкопатия.
Болезнь Шильдера
Син.: Лейкоэнцефалит диффузный периаксиллярный подострый. Редкое прогрессирующее демиелинизирующее заболевание головного мозга, проявляющееся в детском возрасте или у взрослых. Характеризуется поражением белого вещества больших полушарий, формированием в них чаще симметричных обширных (2х2 см и больше) очагов области семиовального центра. Дебютирует головной болью, небольшими подъемами температуры, недомоганием. В дальнейшем – хореоатетоидные гиперкинезы, судорожные припадки, спастические парезы или параличи, псевдобульбарные, мозжечковые, тазовые расстройства, гемианопсия, корковая слепота, межъядерная офтальмоплегия, расстройство обоняния, слуха, высших корковых функций. Когнитивные расстройства, прогрессирующая деменция. Течение подострое или хроническое. Описал в 1912 г. немецкий психиатр Schilder.
Шильдера болезнь
Schilder, 1912) – редкое заболевание неизвестной этиологии, обусловленное прогрессирующим демиелинизирующим процессом в веществе головного мозга (полушария, мозжечок). Начало заболевания относится к молодому и детскому, реже более позднему возрасту. Характерными признаками заболевания являются: 1. нарастающая деменция; 2. расстройства речи, зрения и слуха (до глухоты и амавроза); 3. эпилептические припадки; 4. очаговая неврологическая патология; 5. спастические параличи; 6. псевдобульбарная симптоматика. Спустя 2-3 года пациенты погибают при явлениях кахексии и децеребрационной ригидности. Предполагается близость заболевания к рассеянному склерозу. Синонимы: Диффузный склероз нервной системы, Прогрессирующая мозговая лейкопатия, Диффузный прогрессирующий склеротический периаксиальный энцефалит, Прогрессирующая склеротическая энцефалолейкопатия.
Д.В. Иванов,
Николаевская областная психиатрическая больница № 1
Болезнь Шильдера, или диффузный периаксиальный энцефалит Шильдера (далее ДПЭ) – редкое, неуклонно прогрессирующее заболевание нервной системы, характеризующееся формированием патологических очагов демиелинизации в белом веществе головного мозга. Вопрос возрастного дебюта заболевания остается спорным и имеет неоднозначные данные. Зарубежные авторы в своих наблюдениях подчеркивают, что для ДПЭ характерно начало в детском возрасте (7-12 лет) . Однако ряд отечественных ученых настаивают, что частота заболеваемости не зависит от возрастной категории и отмечаются одинаково часто как у детей, так и у взрослых .
Этиология ДПЭ остается неизвестной. Патоморфологический субстрат – очаги обширной демиелинизации в белом веществе обоих полушарий – часто асимметричные, с четко очерченными и заостренными краями . Также участки демиелинизации могут первично встречаться в мозжечке и стволе мозга. Некоторые авторы описывают случаи, когда наряду с основными, большими очагами, встречаются участки несколько меньшего размера, округлой формы, напоминающие бляшковидные участки демиелинизации при рассеянном склерозе. Их появление более характерно для заболевания, начавшегося в подростковом и зрелом возрасте .
При патогистологическом исследовании выявляются участки фибриллярного глиоза с гигантскими многоядерными астроцитами и периваскулярная инфильтрация плазматическими клетками .
Клиническая картина болезни Шильдера полиморфна и неспецифична . Выделяют следующие основные группы симптомов:
Психические расстройства, сопровождающиеся нарушением поведения по апато-абулическому типу, а также когнитивным снижением, вплоть до тотальной деменции;
Поражение черепно-мозговых нервов (глухота, офтальмоплегия, парез лицевого нерва, бульбарный синдром, оптический неврит);
Поражение мозжечка (нистагм, интенционный тремор, скандированная речь, атаксия);
Поражение зрительной коры (корковая слепота, гемианопсия);
Судорожный синдром (чаще всего не сопровождающийся специфическими изменениями на ЭЭГ);
Экстрапирамидные нарушения;
Общемозговые симптомы.
Прижизненная диагностика ДПЭ требует тщательного анализа данных и дифференцирования от ряда других клинически схожих заболеваний. Основным методом диагностики является МРТ-исследование головного мозга, которое должно показать наличие одного большого или двух сливающихся между собой участков демиелинизации в белом веществе головного мозга, чаще всего расположенных перивентрикулярно, билатерально или монолатерально . ЭЭГ-данные при ДПЭ неспецифичны и проявляются в виде дезорганизации волн и снижения α-активности, что свидетельствует о диффузном поражении головного мозга . Возможное наличие латерализованных эпилептиформных разрядов (PLEDS) указывает на развитие подострого склерозирующего энцефалита, особенно если дебют заболевания приходится на детский возраст.
Для окончательной постановки диагноза ДПЭ необходимо руководствоваться клиническими критериями, разработанными C.M. Poser в 1985 г., которые представлены далее.
1. Один или два очага округлой формы, расположенные симметрично в каждом полушарии, преимущественно в семиовальном центре. Размер очагов составляет не менее 2 × 3 см.
2. Отсутствие клинических или лабораторных данных относительно патологии надпочечников.
3. Сывороточная концентрация жирных кислот с длинной цепью в пределах физиологической нормы.
4. Отсутствие любых других поражений головного мозга, определяющихся клинически, лабораторно или инструментально.
5. Отсутствие патологии со стороны периферической нервной системы.
6. Наличие очагов диффузного хронического склероза на аутопсии.
Прогноз при ДПЭ неблагоприятный, заболевание приводит к быстрой инвалидизации и смерти. Специфических методов лечения не разработано. Рекомендовано назначение метилпреднизолона в дозе 20-30 мг/кг массы тела парентерально, с последующим переводом на пероральный прием .
Представляем вашему вниманию клинический случай, когда больная была доставлена на лечение в психиатрический стационар в связи с выраженными поведенческими и когнитивными нарушениями.
Клинический случай
Анамнез жизни и заболевания
Больная П., 1952 г.р., родилась от первой беременности в срок, развитие раннее, без особенностей. Получила высшее образование по специальности «биохимия». Работала учителем биологии, затем – врачом-лаборантом. Проживает одна, родных братьев и сестер не имеет, родители умерли, мать – от сахарного диабета, а отец – от онкологического заболевания. Анамнез психическими заболеваниями не отягощен. Наличие черепно-мозговой травмы, употребление алкоголя и психоактивных веществ в анамнезе отсутствуют. Хроническими заболеваниями не страдает.
В психиатрический стационар поступила 27.11.2014 г. Ранее в поле зрения психиатров не попадала. По характеру всегда была замкнутой, обособленной. Первые симптомы заболевания возникли около двух лет назад и были замечены сотрудниками на работе (в клинической лаборатории). Стала допускать ошибки при работе с аппаратами и реактивами, перестала справляться с эксплуатацией оборудования, нарушился почерк, не могла писать слова в одну строку. Появились неуверенность при ходьбе, неловкость движений, ударялась о дверные косяки и задевала мебель при ходьбе, отмечались эпизоды падения без внешних причин.
Одновременно с этим изменилось поведение больной: стала подозрительной, считала, что ее хотят «подсидеть» на работе, что сотрудники планируют забрать у нее квартиру. Появилась неряшливость, неаккуратность во внешнем виде: приходила на работу неопрятной, в грязной одежде, порой надетой наизнанку. На любые замечания со стороны коллег реагировала обидой и сразу замыкалась в себе. От предложенного обследования у невропатолога и окулиста пациентка отказалась, отреагировала негативно и уволилась с работы несмотря на то что продолжала оставаться на хорошем счету у коллег.
После увольнения больная вела крайне обособленный образ жизни, из квартиры выходила очень редко, в магазин или к соседям. Без объективных причин выбросила из квартиры много ценных вещей, оставшихся от родителей. Приютила у себя дома двух бездомных собак и кота, которых не кормила и не выводила на улицу – они справляли нужду в квартире. Животные ходили изголодавшиеся, пока собаки не разорвали кота и не съели его, что не вызвало никакой реакции у пациентки.
Все это время больная находилась под периодическим присмотром родственницы, которая приносила ей еду. Пациентка зачастую выносила ее на улицу, а примерно за месяц до госпитализации и вовсе отказалась от приема пищи. В общей сложности за последний год она похудела приблизительно на 30 кг.
При поступлении состояние больной средней степени тяжести. Наблюдаются сильный запах ацетона изо рта, повторная рвота.
Психический статус
Сознание больной не помрачено. Контакту доступна крайне формально. Аллопсихически ориентирована верно, аутопсихически дезориентирована. На большинство вопросов отвечать отказывается, утверждает, что ей все неприятны, и она никого не хочет видеть. Указывая на сопровождающую ее родственницу, говорит: «Эта женщина хочет забрать мою квартиру, но кто она такая, я не знаю». Галлюцинаторная симптоматика не обнаружена. Интеллектуально-мнестические функции резко снижены. Внешне крайне неопрятна, неряшлива, обнаружен педикулез. К своему внешнему виду равнодушна, к состоянию некритична.
Данные патопсихологического исследования
У пациентки наблюдаются нарушение цветового гнозиса, оптико-пространственная агнозия, нарушение конструктивного праксиса, признаки акалькулии. Провести более детальное исследование не представляется возможным в силу ее психического состояния.
Неврологический статус
Больная в позе Ромберга неустойчива, пальценосовую пробу выполняет с мимопопаданием. Выявлен выраженный интенционный тремор верхних конечностей. Походка с элементами атаксии по мозжечковому типу. Сухожильные рефлексы живые. Отмечается ряд нижних патологических рефлексов: Штрюмпеля, Россолимо, Жуковского – Корнилова, Оппенгейма, Бабинского, а также хватательный рефлекс. Наблюдаются офтальмоплегия, паралич аккомодации.
При офтальмологическом исследовании определяется преходящая гомонимная гемианопсия, границы диска зрительного нерва смазаны, наблюдается легкая гиперемия.
Результаты обследования
При МРТ-исследовании головного мозга пациентки (аппарат Philips Interna 1,5 TI) на фоне атрофических изменений вещества головного мозга субкортикально, перивентрикулярно в белом веществе обеих гемисфер отмечены гиперинтенсивные очаговые изменения на Т2-ВИ и в режиме FLAIR (размер – до 4 мм); у заднего рога латерального желудочка справа наблюдается участок глиоза (размер – 9,7 × 29,8 × 18,7 мм). Расширены периваскулярные пространства. Субарахноидальное пространство больших полушарий заместительно расширено на всем протяжении.
Наблюдение в динамике
Психическое и неврологическое состояние больной продолжало прогредиентно ухудшаться, отличаясь выраженной лабильностью симптоматики. Усиливались явления атаксии и кинестетической апраксии. Пациентка не могла передвигаться без посторонней поддержки, пищу принимала из рук медперсонала. Часто отмечались речевые и двигательные стереотипии, продолжавшиеся на протяжении всего дня. Трижды наблюдались тонические постуральные судорожные припадки. Нарастала грубая дезориентировка в месте и собственной личности. Физиологические отправления контролировались не всегда, часто нуждалась в напоминании о необходимости сходить в туалет. Бредовые идеи о том, что у больной хотят отнять квартиру, поначалу занимавшие ее, постепенно редуцировались. Мышление становилось все более аморфным, но на простые вопросы могла дать достаточно адекватный ответ. К своему состоянию оставалась некритична.
Обоснование диагноза и обсуждение
Как видно из приведенных данных, заболевание у пациентки дебютировало практически одномоментным нарушением со стороны неврологической и психической сфер. Отмечалось заострение преморбидных черт личности с последующим искажением и развитием апато-абулического симптомокомплекса. Следует подчеркнуть, что быстро прогрессирующее когнитивное снижение с переходом в тотальную деменцию является неспецифическим проявлением и может наблюдаться и при других патологиях головного мозга. Однако наличие массивного очага глиоза, множественных мелких участков демиелинизации, вовлечение в процесс пирамидной системы, мозжечка и стволовых структур свидетельствуют о наличии ДПЭ.
Клиническая симптоматика, наблюдаемая у пациентки, укладывается в вышеуказанные критерии Posner, за исключением прижизненной биопсии, которая не проводилась.
Данные МРТ-диагностики напоминают изменения при рассеянном склерозе, фульминантная форма которого также может привести к быстрому прогрессированию психического и неврологического дефицита. Но наличие большого очага глиоза является атипичным для рассеянного склероза и более характерно для ДПЭ . Схожая симптоматика и МРТ-картина могут наблюдаться при адренолейкодистрофии, но никаких объективных данных, указывающих на патологию надпочечников, у больной не выявлено. Отличие от острого диссеминированного энцефаломиелита заключается в отсутствии данных о перенесенном инфекционном заболевании или предшествующей вакцинации, а также в интактности периферического нейрона и отсутствии тетра- и парапарезов.
Больной был выставлен клинический диагноз согласно МКБ-10: деменция при других уточненных болезнях, классифицированных в других рубриках – диффузный периаксиальный энцефалит Шильдера.
Лечение
Пациентке был назначен преднизолон в стартовой дозе 20 мг/кг/сут. Однако на протяжении трех дней от начала терапии у больной отмечалось не свойственное для нее повышение артериального давления, из-за чего дозировка была снижена до 10 мг/кг/сут. Помимо этого, пациентка прошла курс нейропротекторной, сосудистой и антиоксидантной терапии. Несмотря на проводимое лечение, ее психоневрологическое состояние не улучшалось, а продолжало неуклонно ухудшаться.
Выводы
Больная попала в поле зрения врачей лишь спустя два года после начала заболевания, когда терапевтические методы лечения уже неэффективны. Это лишний раз подчеркивает необходимость совершенствования скрининговой системы, под особым прицелом которой должны быть одинокие люди, в том числе пожилого и старческого возраста.
В данном случае психические нарушения являются вторичными по отношению к основному неврологическому заболеванию, а потому такие больные нечасто попадают в поле зрения врача-психиатра. Однако нередко бывают случаи, когда соматический недуг манифестирует с расстройства психических функций, что указывает на необходимость сотрудничества специалистов узкого профиля, особенно по смежным специальностям.
МРТ-диагностика является одним из ведущих методов исследования не только в практике врачей-интернистов, но и в психиатрической отрасли. Это свидетельствует о важности создания условий для доступа к данному исследованию как можно более широкого контингента лиц, страдающих психической патологией, с целью своевременной диагностики и более эффективного лечения.
Литература
- Марков Д.А., Леонович А.Л. Рассеянный склероз. – М.: Медицина, 1976. – 296 с.
- Линьков В.В., Андреев А.Г., Лебедева Л.В., Виноградов В.В., Кустова И.Р., Гаранина Е.С. Лейкоэнцефалит Шильдера. Случай из практики // Вестник Ивановской медицинской академии. – 2012. – Т. 17, № 1. – С. 68-70.
- Мельничук П.В. Болезни нервной системы (руководство для врачей). – М.: Медицина, 1982. – 368 с.
- Йорданов Б., Янков Я. Редкие синдромы и заболевания нервной системы. – М.: Медицина, 1981.
- Мартынов Ю.С. Лекции по невропатологии. – Москва, 1969. – 260 с.
- Dresser L.P., Tourian A.Y., Anthony D.C. A case of myelinoclastic diffuse sclerosis in an adult // Department of Medicine, Division of Neurology, and the Department of Pathology, Duke University Medical Center, Durham, NC. – 1991. – V. 41, № 2 – P. 1.
- Christensen E., Fog M. A case of Schilder’s disease in an adult with remarks to the etiology and pathogenesis // Acta Psychiatrica Scandinavica. – 2007. – V. 30. – P. 141-154.
- Rust R.S. Jr., Nowack W.J, Talavera F., Benbadis S.R. Diffuse Sclerosis Treatment & Management // Medscape.
- Miller Neil R., Newman Nancy J., Biousse, Valerie, Kerrison, Walsh J.B. Hoyt’s Clinical Neuro-Ophthalmology / Sixth edition. – North American Neuro-Ophthalmology Association, 2005. – 3573 p.
- Gupta R.K., Kumar S. Magnetic Resonance Imaging of Neurological Diseases in Tropics / First edition. – Jaypee Brothers Medical Publishers (P) Ltd, 2014. – 344 p.
- Kim J.H., Lee S.M., Kim H.D., Lee J.S., Kang H. Schilder’s disease in a young child with tumefactive demyelinating brain lesion // Neurology Asia. – 2013. – V. 18 (4). – P. 419-421.
Лейкоэнцефалит (ЛЭ) Шильдера
- диффузный склероз описан автором в 1912 г. Заболевание характеризуется тяжелой двусторонней демиелинизацией больших полушарий и ствола мозга (при довольно сохранных осевых цилиндрах), выраженной глиальной и п ер ив аокул ярной воспалительной реакцией. Морфологическая картина и особенности течения заболевания позволяют отнести его к группе рассеянного склероза.
Лейкоэнцефалит Шильдера одинаково часто встречается как в детском, так и во взрослом возрасте. Начало заболевания обычно постепенное, очень редко инсультообразное. Первыми основными проявлениями лейкоэнцефалита могут быть изменения поведения, прогрессирующие нарушения высших психических функций (зрительного и слухового гнозиса, праксиса, речи, интеллекта), эпилептические припадки, психотические состояния, пирамидные парезы. «Типичной» картины ЛЭ не существует. В одних случаях болезнь начинается под маской опухоли головного мозга, в других - протекает как психическое заболевание, в третьих - напоминает рассеянный склероз. Такое многообразие клинических проявлений обусловлено диффузным характером демиелинизирующего процесса в больших полушариях головного мозга, величиной очагов демиелинизации, а также степенью выраженности периваскулярного отека.
Наиболее частой и типичной формой течения лейкоэнцефалита Шильдера является псевдотуморозная. При ней признаки внутричерепной гипертензии (головные боли со рвотой, прогрессирующие застойные изменения дисков зрительных нервов) сопровождают нарастающую, преимущественно однофокусную симптоматику (джексоновские припадки, пирамидный гемипарез, центральная гомонимная гемианопсия).
Нередко при этом выявляются очаговая патологическая активность на ЭЭГ, небольшое повышение давления спинномозговой жидкости, белково-клеточная диссоциация (белок от 0,7 до 3,3%о при нормальном цитозе).
Особенностями псевдотуморозной формы ЛЭ по сравнению с опухолевым процессом являются: наличие признаков многоочаговости и двусторонности поражения; тенденция к колебаниям выраженности симптомов и ремиссиям, типичная для демиелинизирующего процесса; диссоциация между застойными изменениями дисков зрительных нервов и отсутствием повышения давления спинномозговой жидкости и гипертензивных изменений на жраниаграмме; в жидкости значительная гипергаммаглобулинорахия, частые патологические изменения реакции Ланге; на ЭЭГ - ранние грубые диффузные изменения; отсутствие смещения М-эха на эхоЭГ; при ремиссии - уменьшение белково-клеточной диссоциации в спинномозговой жидкости, очаговых изменений на ЭЭГ.
Комплексная терапия должна быть направлена, как и при лечении рассеянного склероза, прежде всего на подавление аутоаллергических реакций и включать гормоны коры надпочечников, а также десенсибилизирующие средства. Среди симптоматических средств наиболее важными являются противосудорожные и миотонолитические.
Лейкоэнцефалит представляет собой форму вирусного происхождения. При лейкоэнцефалите преимущественно поражается белое вещество мозга. Помимо этого присутствуют дистрофические изменения в нейронах.
Предположительно заболевание имеет аллергическо-вирусную природу. На данный момент рассматриваются версии о возможном происхождении болезни от вирусов: кори, бешенства, вируса герпеса 3 типа.
Выделяют три основные клинико-морфологичекие формы лейкоэнцефалита:
- подострый склерозирующий Ван-Богарта;
- периаксиальный Шильдера;
- острый геморрагический.
При этом исследование пораженного энцефалитом мозга дает следующее результаты: наблюдается атрофия извилин и расширение борозд, участки димиелинизации обнаруживаются во всех отделах мозга, в основном в белом веществе, но также и в сером веществе коры. Наблюдается умеренное расширение желудочков.

Провоцирующие факторы
Существует предположение, что лейкоэнцнфалит Ван-Богарта вызывается вирусом кори, который находился в латентном состоянии в нейронах мозга и активизировался при определенных процессах.
Наиболее часто встречающиеся формы — лейкоэнцефалиты Шильдера и Ван-Богарта. Синдром Ван-Богарта сопровождается экстрапирамидными симптомами, эпилептиформными припадками, слабоумием.
Для болезни Шильдера характерны нарушения двигательной системы, зрения или ретробульбарный нефрит.
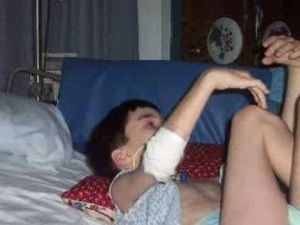 Симптомы появляются внезапно, для них характерен неуклонный прогресс. На первых порах болезнь сопровождается психическими нарушениями. У маленьких детей это проявляется в виде раздражительности, апатии, агрессивности, вялости.
Симптомы появляются внезапно, для них характерен неуклонный прогресс. На первых порах болезнь сопровождается психическими нарушениями. У маленьких детей это проявляется в виде раздражительности, апатии, агрессивности, вялости.
Постепенно начинают пропадать логика мышления, навыки опрятности, снижается словарный запас. На фоне этого в течение нескольких месяцев начинает развиваться неврологическая симптоматика.
Она может включать галлюцинации, повышение , агнозию, апраксию, полиморфные гиперкинезы. После присоединяются нарушения двигательной системы: и параличи, неустойчивая походка, нарушение координации.
Постоянным симптомом является , после присоединяются трофические и вегетативные расстройства.
Симптоматика каждой формы заболевания
При заболевании Ван-Бограрта чаще, чем при других формах, отмечается и ствола головного мозга. Для этой формы лейкоэнцефалита характерны ранние проявление экстрапирамидных расстройств, пирамидные симптомы присоединяются на поздней стадии. При этой форме болезни редко встречаются .
При лейкоэнцефалите Шильдера в отличии от других клинических форм и , наблюдается ранняя дистрофия аксонов. При этой форме преобладают пирамидные симптомы, она сопровождается частыми эпилептическими припадками.
При остром геморрагическом энцефалите начало острое, с молниеносным нарастанием симптомов. Заболевают в 20-40 лет при длительности течения до 2 недель. Сопровождается , нарушением сознания, ригидностью мышц шеи, комой.
Для этой формы лейкоэнцефалита характерны судороги и нарушения двигательного аппарата, паралич. В очень редких случаях может развиться подострая или хроническая форма.
Подход к диагностике
На ранней стадии развития болезнь легко перепутать с шизофренией, . Далее проводится дифдиагноз с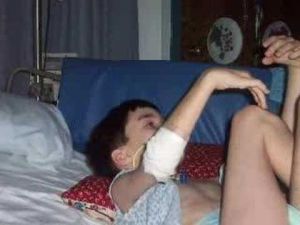 .
.
Учитываются диффузность поражения, наличие или отсутствие повышенного внутричерепного давления, признаки смещения срединных структур мозга на . Для подтверждения диагноза проводится и иммунологические исследования.
Лабораторные исследования оказывают умеренный пиноцитоз в цереброспинальной жидкости, повышения уровня гамма-глобулина и белка. Наблюдаются изменения реакции Ланге. ЭЭГ регистрирует комплексы Радемеккера. Наиболее информативными считаются КТ и МРТ.
Начальные стадии острого склерозирующего энцефалита схожи с шизофренией. В это время отдиффиринцировать ее сложно, но имеется ряд симптомов которые все же дают возможность исключить шизофрению: потеря памяти, быстрая истощаемость при интеллектуальной деятельности.
При лабораторных данных наблюдается повышенное содержание белка и гамма-глобулина.
В целях терапии
 Применяются препараты из группы кортикостероидов. Преднизолон 1-1,5 мг/кг веса, на протяжении 2-3 недель постепенно снижая дозу. Если терапия дает положительный результат, ее продолжаю в поддерживающих дозах. Повышают при эпизодах обострения болезни.
Применяются препараты из группы кортикостероидов. Преднизолон 1-1,5 мг/кг веса, на протяжении 2-3 недель постепенно снижая дозу. Если терапия дает положительный результат, ее продолжаю в поддерживающих дозах. Повышают при эпизодах обострения болезни.
Параллельно назначаются антибиотики: Пенициллин, Эритромицин, Тетрациклин.
Увеличивают прием солей калия, при этом снижая количество солей натрия. При необходимости назначают витамины, а также психотропные препараты.
Прогноз и смертность
Заболевание неуклонно прогрессирует, приводя всегда к летальному исходу. Длительность развития заболевания составляет от 0,5 до 3 лет.
В некоторых случаях возможно дольше, при этом болезнь протекает хронически, периодически наблюдаются ремиссии. Во время ремиссии симптомы заболевания могут полностью отсутствовать.
Реальный клинический случай
Пациент Вадик, 10 лет поступил в больницу с жалобами на плохую учебу и неуспеваемость. Наследственность: фобии у матери, отец здоров.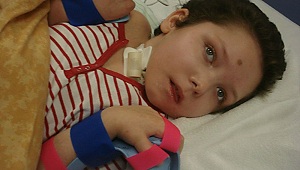
Сестра отца совершила самоубийство. В семье отца есть умственно отсталый родственник. Рожден от третьей беременности, две первые закончились родами раньше срока, дети не выжили.
Третья беременности протекала с обмороками, родился во время. Развивался больной без отклонений, посещал детский сад, часто болел. В 10 лет стал страдать от ночного недержания мочи. Вскоре после этого появились необоснованные страхи, ухудшилась успеваемость. Стал отставать в развитии, гримасничать, стал очень невнимательным.
Результаты обследования: кожа сухая, красноватая, тургор понижен, питается удовлетворительно. Имеется усиленная пигментация на туловище. Периодически диарея, Холестерин 303 мг%, белок в ликворе 0,67. Положительный результат реакции Нонне-Апельта.
Психическое состояние ребенка: нормально поддерживает беседу, богатый набор слов, через некоторое время начинает размахивать руками, вертеть волосы, по-особенному загибать палец. Быстро устает, не сосредотачивается, имеются головные боли.
Заключение эндокринолога: Отсутствуют признаки дисфункция коры надпочечников.
Заключение нейрохирурга: двухсторонний синдром Кернинга, ограничение взора вверх, возможна опухоль в лобной доле.
 Результаты пневмоэнцефалографии: расположение желудочков симметричное, отсутствие опухоли в мозговых полушариях. с изменениями в мозговых оболочках.
Результаты пневмоэнцефалографии: расположение желудочков симметричное, отсутствие опухоли в мозговых полушариях. с изменениями в мозговых оболочках.
Проведено лечение Пенициллином и Бийохинолом. После этого мальчик выписан с диагнозом: неясный процесс в лобных долях.
Через пару месяцев повторно поступил на лечение.
При поступлении: расстройство конвергенции глаза, сглаженность левой носогубной складки. Снижение рефлексов кремастерных и брюшных. Наличие белка в ликворе 1,32.
При обследовании: резкие нарушения психики, быстрая интеллектуальная утомляемость. С нарушениями, но сохранены навыки чтения и письма. Отмечается безопасность на свидании с родителями. Диагноз — лейкоэнцефалит.
В дальнейшем заболевание прогрессирует от начала болезни до появления эпилептических припадков через 24 месяца. После госпитализации через год перестал вставать. Последние месяцы полностью беспомощен, жалобно плакал. Скончался при падениях сердечной деятельности.
Болезнь шильдера
Лейкоэнцефалит Шильдера (диффузный склероз) описан в 1912 г. Заболевание характеризуется тяжелой двусторонней демиелинизацией полушарий большого мозга и мозгового ствола (при довольно сохранных осевых цилиндрах), выраженной глиальной и периваскулярной воспалительной реакцией. Морфологическая картина и особенности течения заболевания позволяют отнести его к группе демиелинизирующих заболеваний.
Лейкоэнцефалит одинаково часто встречается как у детей, так и взрослых. Начало заболевания обычно постепенное, очень редко инсультообразное. Первыми основными проявлениями лейкоэнцефалита могут быть изменения поведения, прогрессирующие нарушения высших психических функций (зрительного и слухового гнозиса, праксиса, речи, интеллекта), эпилептические припадки, психотические состояния, пирамидные парезы. “Типичной” картины лейкоэнцефалита не существует. В одних случаях болезнь начинается под маской опухоли головного мозга, в других протекает как психическое заболевание, в третьих напоминает рассеянный склероз. Такое многообразие клинических проявлений обусловлено диффузным характером демиелинизирующего процесса в полушариях большого мозга, величиной очагов демиелинизации, а также степенью выраженности периваскулярного отека.
Наиболее частой и типичной формой течения лейкоэнцефалита является псевдотуморозная форма. При ней признаки внутричерепной гипертензии (головные боли с рвотой, прогрессирующие застойные изменения дисков зрительных нервов) сопровождают нарастающую преимущественно однофокусную симптоматику (джексоновские припадки, пирамидный гемипарез, центральная гомонимная гемианопсия). Нередко при этом выявляются очаговая патологическая активность на ЭЭГ, небольшое повышенное давление цереброспинальной жидкости, белково-клеточная диссоциация (белок 0,7-3,3 г/л при нормальном цитозе).
Особенностями псевдотуморозной формы лейкоэнцефалита по сравнению с опухолевым процессом являются наличие признаков многоочаговости и двусторонности поражения; тенденция к колебаниям выраженности симптомов и ремиссиям, типичная для демиелинизирущего процесса; диссоциация между застойными изменениями дисков зрительных нервов и отсутствием повышения давления цереброспинальной жидкости и гипертензивных изменений на краниограмме; в жидкости значительная гипергаммаглобулинорахия, частые патологические изменения реакции Ланге; на ЭЭГ - ранние грубые диффузные изменения; при ремиссии - уменьшение белково-клеточной диссоциации в цереброспинальной жидкости, очаговых изменений на ЭЭГ. Исключительно важное диагностическое значение имеют результаты КТ и особенно МРТ.
Комплексная терапия должна быть направлена, как и при лечении рассеянного склероза, прежде всего на подавление аутоиммунных реакций и включать в себя гормоны коры надпочечников. Среди симптоматических средств наиболее важными являются противосудорожные и миотонолитические.
Острая воспалительная демиелинизирующая полирадикулоневропатия Гийена - Барре (ОВДП)
В 1916 г. Гийен, Барре и Штроль описали острый периферический паралич с белково-клеточной диссоциацией в цереброспиналыюй жидкости и благоприятным прогнозом. Описанная ими клиническая картина практически не отличалась от «острого восходящего паралича», описанного Ландри еще в 1895 г. ОВДП встречается с частотой 1,7 ва 100 000 населения, равномерно в разных регионах, в любом возрасте, у мужчин чаще, чем у женщин. В настоящее время ОВДП является наиболее частой причиной развития острого периферического паралича наряду с острым полимиозитом и миастенией. Этиология заболевания неизвестна, иногда оно ассоциируется с банальными инфекциями. Обнаружение в сыворотке больных антител к миелину ПНС, а также развитие сегментарной демиелинизации после введения сыворотки в седалищный нерв крысы (экспериментальный аллергический неврит) убедительно свидетельствуют о том, что в основе патогенеза заболевания лежат иммунологические нарушения. Основным местом иммунного конфликта является субпериневральное пространство. На фоне иммунных нарушений возникают отек, воспалительно-клеточная инфильтрация и диффузная первичная сегментарная демиелинизация в первую очередь в передних корешках и проксимальных отделах спинномозговых нервов, сплетениях, нервах конечностей и вегетативных узлах.
Примерно у половины больных за 1-3 нед до появления первых неврологических симптомов наблюдаются заболевания верхних дыхательных путей, преходящие острые кишечные расстройства, ангина. У 50% в начале заболевания встречаются парестезия в стопах, миалгии в ногах, у 20% - сенсомоторные нарушения в дистальных отделах конечностей, у 20% - только слабость, нередко - краниальная невропатия (двусторонний парез мимических мышц, бульбарные и глазодвигательные нарушения).
Ведущим симптомом являются вялые параличи. Мышцы, как правило, поражаются диффузно и симметрично. Мышечная слабость чаще распространяется в восходящем направлении, захватывая мышцы ног и тазового пояса, туловища, шеи, дыхательную мускулатуру. Мышечная слабость прогрессирует обычно в течение 2-3 нед (средний срок 7-15 дней), но иногда тетраплегия может развиться в течение нескольких часов или суток. В первые дни болезни часто наблюдаются миалгии, обусловленные, вероятно, миозитическим процессом, поскольку сопровождаются увеличением мышечных аминотрансфераз. Миалгии обычно стихают без лечения через неделю. При прогрессировании заболевания могут развиться дыхательная недостаточность и бульбарные нарушения, в связи с чем необходим перевод больных на ИВЛ и зондовое кормление. Перевод больных на ИВЛ проводится при жизненной емкости легких менее 15 мл/кг. Поражение диафрагмального нерва приводит к ограничению экскурсии диафрагмы и парадоксальному типу брюшного дыхания (втягивание передней брюшной стенки при вдохе). У многих больных в острой фазе болезни и при тяжелых двигательных расстройствах имеются вегетативные нарушения: ортостатическая гипотензия, тахикардия, пароксизмальная аритмия с изменениями на ЭКГ (депрессия сегмента ST , инверсия Г-волны, удлинение интервала Q-Т). Вовлечение вегетативного аппарата сердца в редких случаях может привести к внезапной его остановке. Дисфункция тазовых органов возможна в острой фазе болезни. Вегетативные расстройства иногда сохраняются и в отдаленном периоде. Острая пандисавтономия, по-видимому, особый вариант ОВДП, при котором селективно поражаются вегетативные волокна.
У всех больных развивается гипотония мышц. Атрофия мышц в острой фазе не наблюдается, однако у ряда больных с тетрапарезом или тетраплегией в восстановительном периоде отмечается похудение мышц проксимальных или дистальных отделов конечностей. Арефлексия или гипорефлексия не связана с тяжестью паралича или атрофией мышц, а зависит от демиелинизации и блока проведения по пораженным корешкам и нервам. Чувствительные нарушения выражены менее тяжело, чем двигательные, и представлены парестезиями, болью, гипалгезией, гиперестезией в дистальных отделах конечностей. В случае вовлечения проприоцептивной чувствительности возникают сенситивная атаксия и стереоанестезии. Симптомы натяжения нервных стволов (Ласега, Нери) остаются длительное время положительными. Стойкие проводниковые расстройства чувствительности исключают диагноз ОВДП. В основе сенсомоторных нарушений при ОВДП лежит сегментарная демиелинизация. Вовлечение черепных нервов наблюдается у половины больных (лицевые, бульбарные и в 10% глазодвигательные). Иногда при большом увеличении содержания белка в цереброспинальной жидкости наблюдается застойный сосок зрительного нерва.
Белково-клеточная диссоциация в цереброспинальной жидкости имеет исключительное диагностическое значение при ОВДП, однако в 1-ю неделю болезни белок может быть нормальным. Корреляция между содержанием белка в жидкости и клинической картиной отсутствует. Обнаружение в цереброспинальной жидкости более 50 клеток в 1 мм всегда должно вызывать сомнение в отношении ОВДП. Очень витка роль электродиагностики. В фазе прогрессирования двигательных нарушений обнаруживается удлинению дистальной (моторной) латенции, снижение скорости проведения по двигательным и чувствительным волокнам, удлинение F-волны, что связано с сегментарной демиелинизацией и блоком проведения. В первые дни болезни электрофизиологические показатели могут быть нормальными (!). Поводом к дифференциальной диагностике служат 3 основных заболевания: синдром Баннварта, дифтерийная и порфирийная невропатии.
Кортикостероидная терапия долгие годы считалась основным способом лечения. Однако проведенные в последнее десятилетие тщательные контролируемые исследования показали, что стероидная терапия не меняет течения заболевания, и даже может способствовать рецидиву болезни. В настоящее время лечение кортикостероидами ОВДП не рекомендуется!
В то же время показана значительная эффективность плазмафереза. За одну процедуру, которую обычно проводят через день, обменивают 1,5-2 л плазмы. Замещающая жидкость состоит из свежезамороженной плазмы, 4% раствора альбумина и плазмозамещающих растворов. Доказана также эффективность внутривенного введения иммуноглобулина.
Для профилактики тромбообразования в иммобилизированных конечностях рекомендуется назначение гепарина по 5000 ЕД подкожно 2 раза в день.
В среднем лечение ОВДП в условиях стационара проводится в течение 2 мес, последующее восстановление двигательных функций наблюдается в течение 1-2 лет. Прогноз у 1/4 больных отличный, однако 2-5 больных умирают, у 10-20% остаются двигательные нарушения разной степени. Восстановление может продолжаться 2 года.
Своевременная диагностика ОВДП и рациональная терапия (плазмаферез, ИВЛ, парентеральное питание, психологическая поддержка, физиотерапия) значительно улучшают прогноз.


